医影在线
标题: V0475:精典病例,有结果 [打印本页]
作者: yangyudong333 时间: 2008-7-22 01:48
标题: V0475:精典病例,有结果
(结果公布:http://www.radida.com/bbs/forum.php?mod=viewthread&tid=42324)
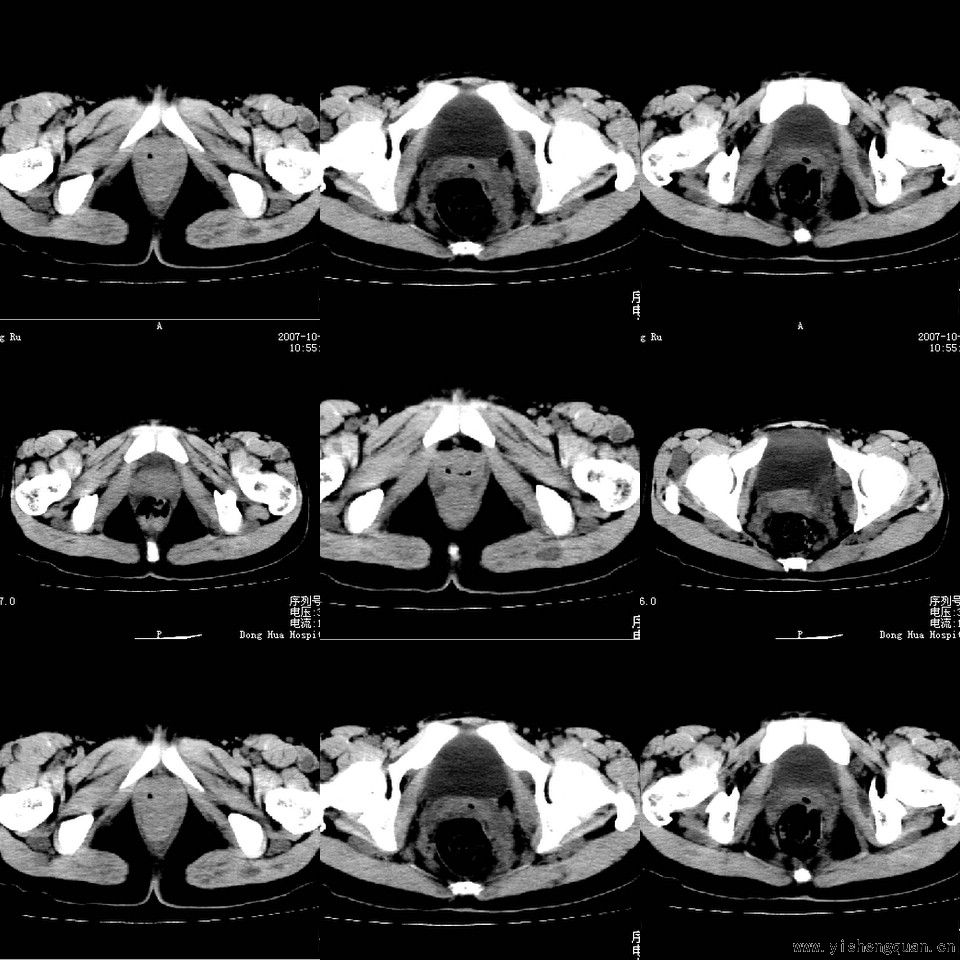
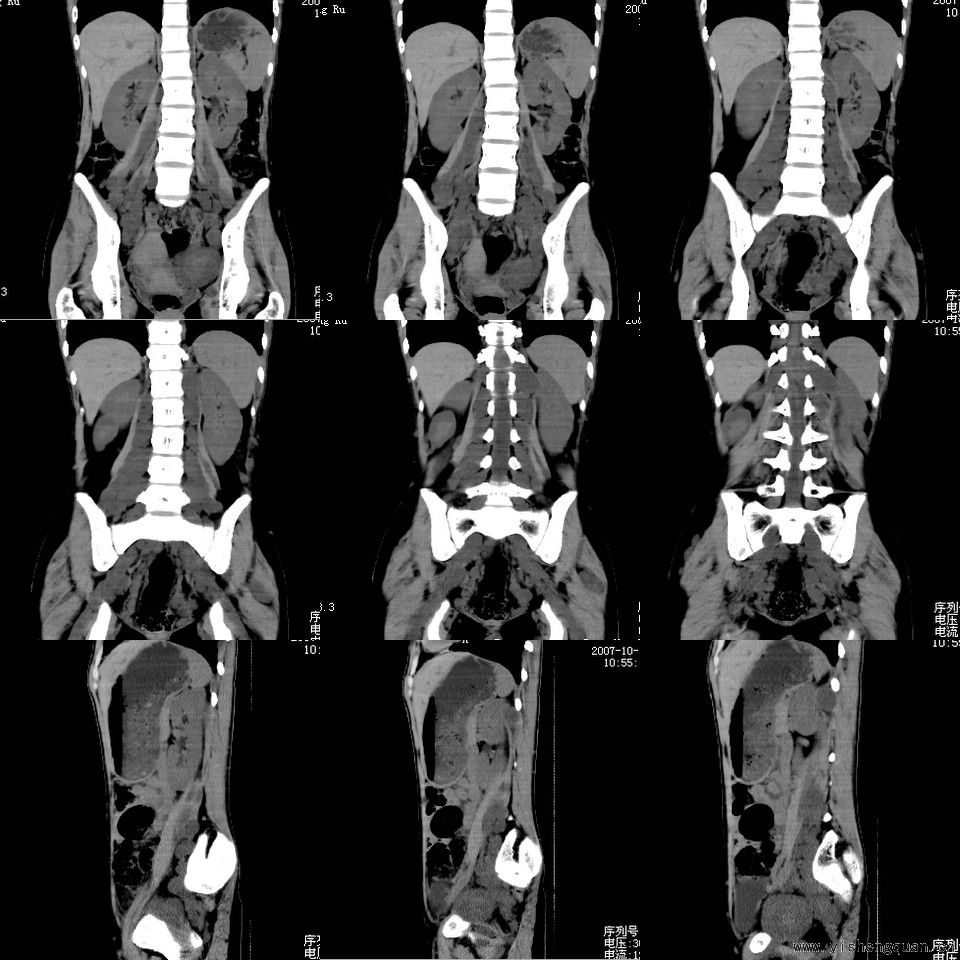
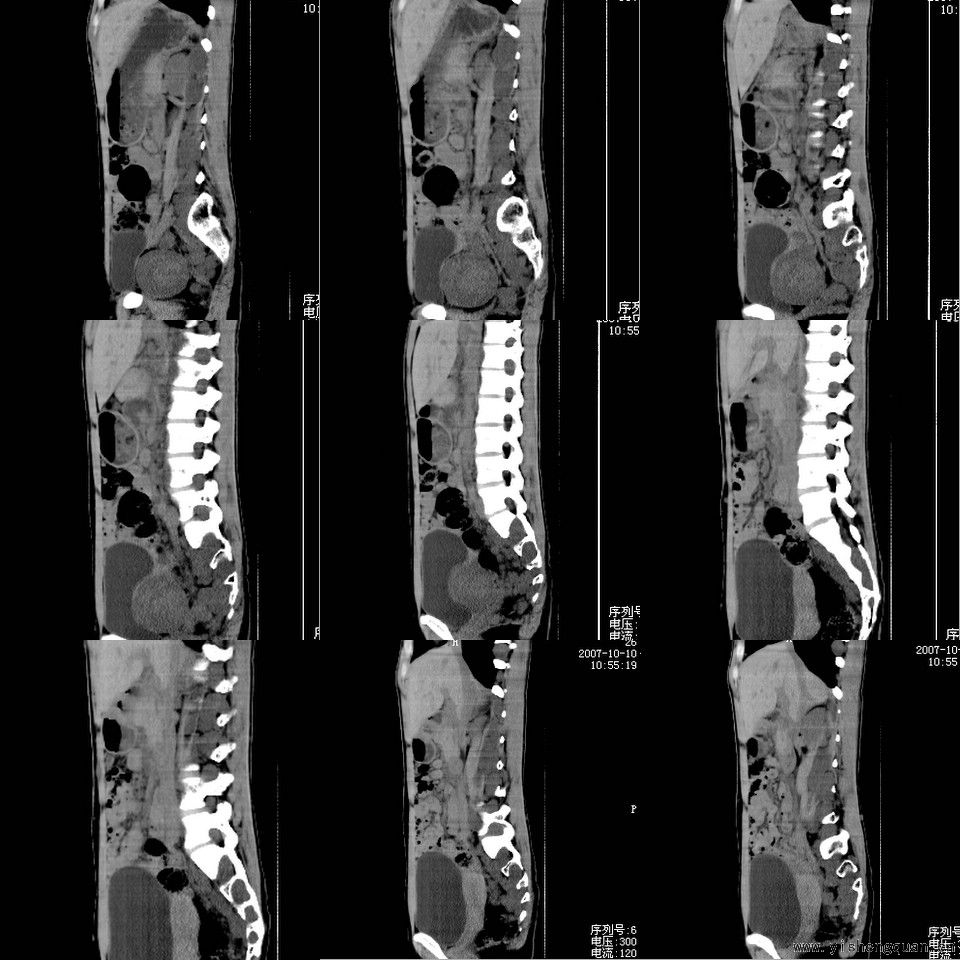
女性,17岁。体检时发现全身触及多个小结节,无触痛.
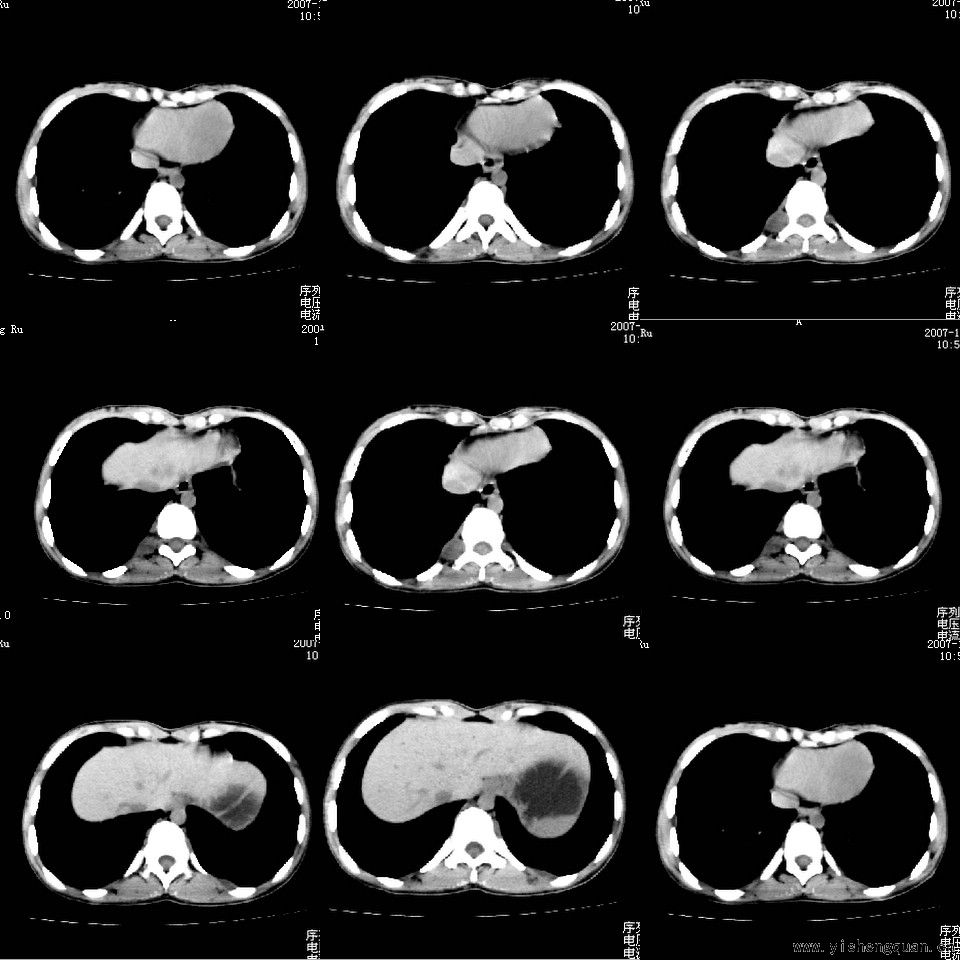
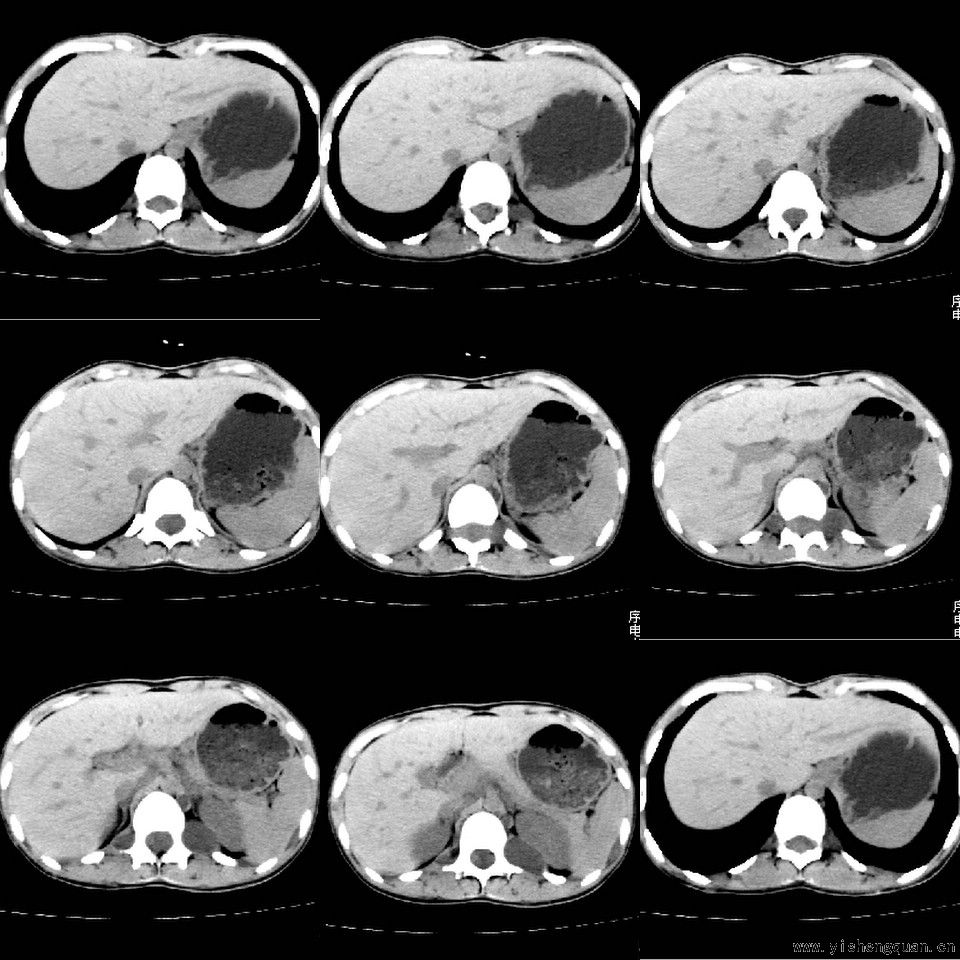
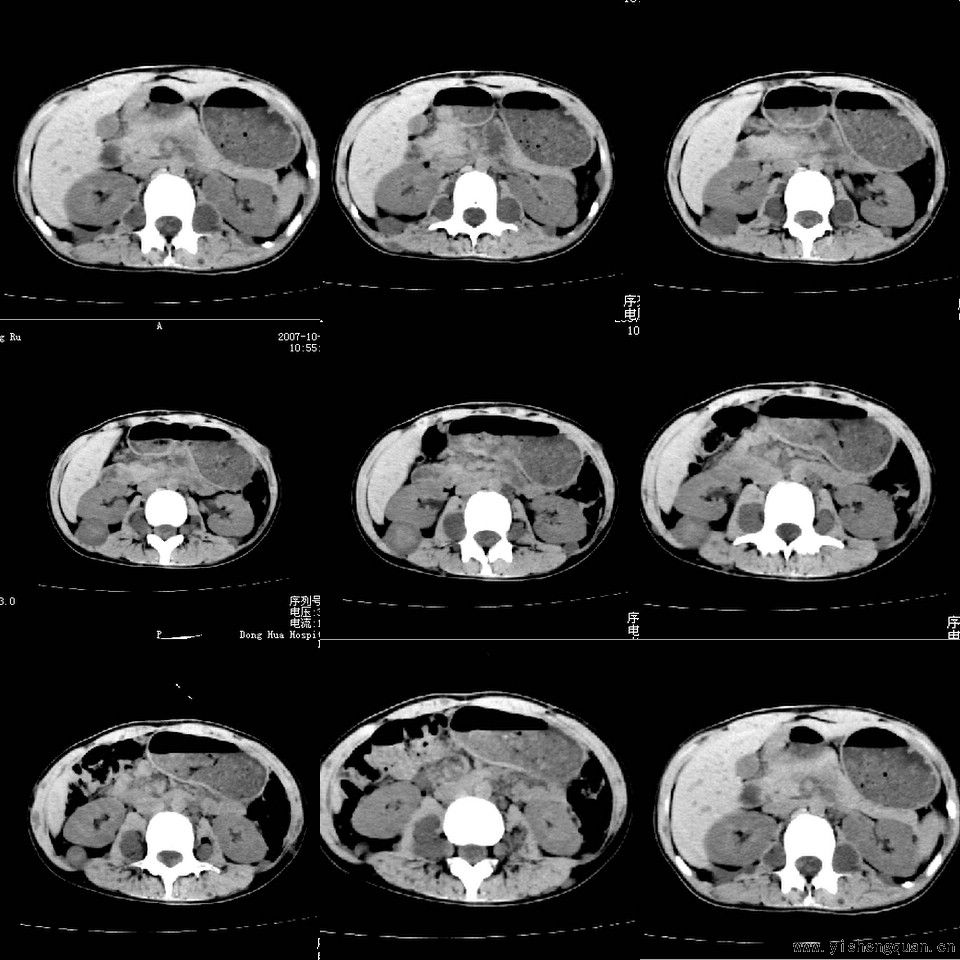
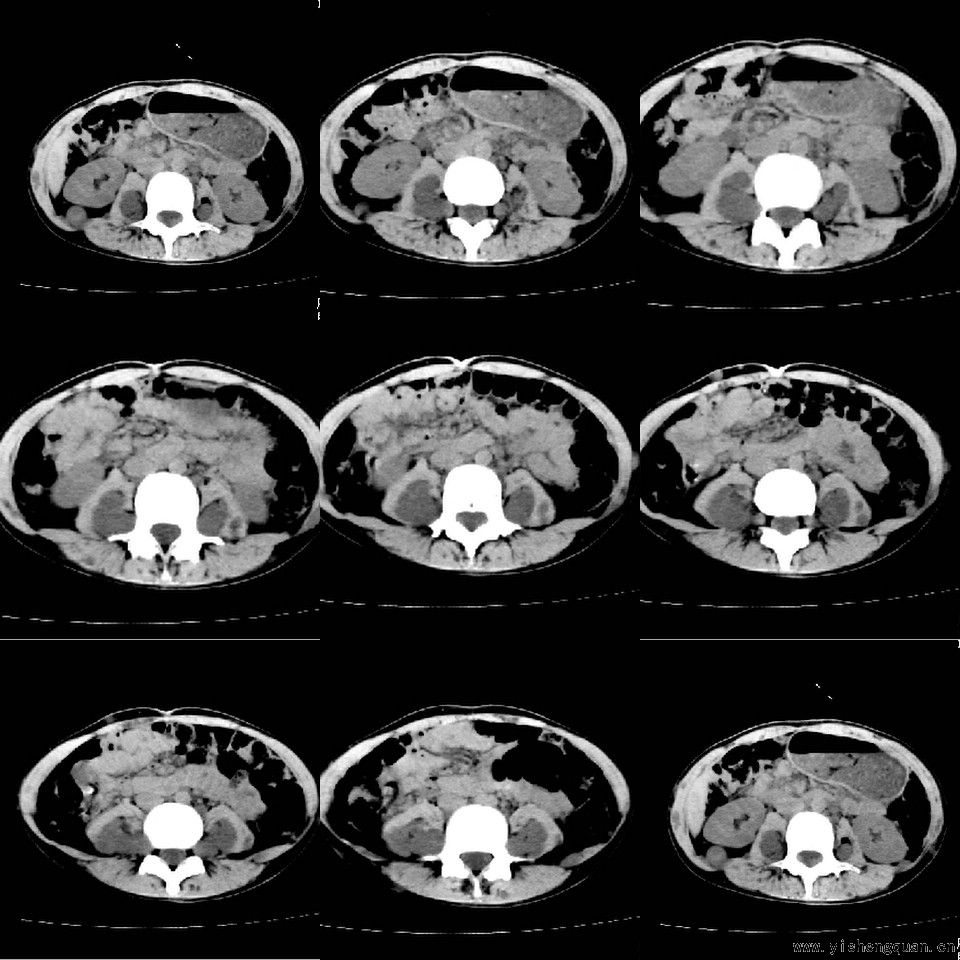




[本贴已被 jiajie 于 2008-7-24 19:28:23 修改过]
[本贴已被 翁志蓬 于 2008-7-31 20:26:33 修改过]
作者: jiajie 时间: 2008-7-22 05:09
标题: 回复:v0475:精典病例,有结果
虽然未提供病史资料,
考虑神经纤维瘤病。


作者: 随光逐影 时间: 2008-7-22 08:19
神经纤维瘤病可能性大。
作者: qc80012345 时间: 2008-7-22 13:01
考虑神经纤维瘤病。
作者: 卜一 时间: 2008-7-22 13:35
多发神经纤维瘤病!
作者: wansheng 时间: 2008-7-22 17:14
多发神经纤维瘤病! 支持!
作者: zhuming943066 时间: 2008-7-22 18:22
附议老师们的意见:多发性神经纤维瘤病
作者: jiangjing 时间: 2008-7-22 19:38
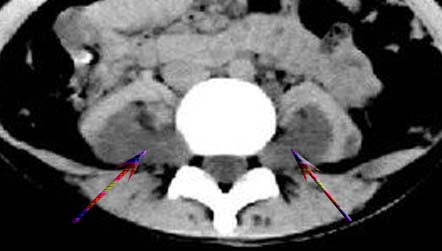
考虑神经纤维瘤病。------脊旁区骶管内多发软组织样结节影,按神经走行方向分布
作者: LIAOQIANG 时间: 2008-7-22 22:23
考虑神经纤维瘤病。
作者: zsl6918 时间: 2008-7-23 00:29
符合多发性神经纤维瘤病。
作者: 同 时间: 2008-7-23 04:32
多发神经纤维瘤病.
作者: LIAIZHI 时间: 2008-7-23 17:14
多发神经纤维瘤病.
作者: yangyudong333 时间: 2008-7-23 20:00
手术病理结果:多发性神经纤维瘤
作者: zjzjr 时间: 2008-7-23 23:25
考虑神经纤维瘤病。
作者: qiuleiyu 时间: 2008-7-24 00:02
符合多发性神经纤维瘤病,建议进一步检查分型。
1、诊断:
①nfi诊断标准(美国nih,1987):6个或6个以上牛奶咖啡斑,青春期前最大直径>5mm,青春期后>15mm;腋窝和腹股沟区雀斑;2个或2个以上神经纤维瘤或丛状神经纤维瘤;一级亲属中有nfi患者;2个或2以上lisch结节;骨损害;
②nfⅱ诊断标准:影像学确诊双侧听神经瘤,一级亲属患nfⅱ伴一侧听神经瘤,或伴神经纤维瘤、脑脊膜瘤、胶质瘤、schwann细胞瘤中的两种,青少年后囊下晶状体浑浊。
作者: 宝天曼 时间: 2008-7-24 07:37
多发神经纤维瘤病! 支持!
作者: wnlyq8688 时间: 2008-7-24 18:26
标题: 回复:v0475:精典病例,有结果
神经纤维瘤病
疾病概述 病因 症状 诊断 治疗 并发症
疾病名称
神经纤维瘤病
疾病概述
神经纤维瘤病是一种少见的遗传性疾病,其特征是皮肤色素沈着斑和多发性神经纤维瘤。约25-50%患者有阳性家族史。患者可在生后不久皮肤即出现色素沈着斑,呈牛奶咖啡色,逐渐增多或扩大。有时皮损出现较迟,在发育期才开始发病,生理变化如发育、妊娠、经绝期、传染病、精神刺激等均可使病情加重。病程缓慢,但到20-50岁时可发生恶变。本病见于世界各地,无性别、年龄和种族差异。 症状体症:1.几乎所有患者都有皮肤色素斑;2.多发性皮肤结节;3.口控损害:表现为乳头状瘤或单侧性巨舌;4.内脏损害:由于颅内肿瘤,血管畸形或骨骼畸形,可引起智力减退、癫痫等神经系统症状。 治疗原则: 一般不需要治疗,除非严重影响美容和功能或有恶变时,才考虑手术治疗。1.对症治疗:如抗癫痫、止痛剂;2.手术治疗:适于皮损严重妨碍美容,或肿瘤太大影响功能,或有疼痛并疑恶变等。必要时进行神经外科手术,切除疼痛的病源。
病因
nf i基因组跨度350kb,cdna长11kb,含59个外显,编码2818个氨基酸,组成32kd的神经纤维素蛋白,分布在神经元。nf i基因是一肿瘤抑制基因,发生易位、缺失、重排或点突变时肿瘤抑制功能丧失而致病。nf ⅱ基因缺失突变引起schwann细胞瘤和脑膜瘤。
症状
神经纤维瘤病为常染色体显性遗传病,是基因缺陷使神经嵴细胞发育异常导致多系统损害。根据临床表现和基因定位分为神经纤维瘤病i型(nf i)和ⅱ型(nf ⅱ)。nf i由von recklinghausen(1982)首次描述,主要特征为皮肤牛奶咖啡斑和周围神经多发性神经纤维瘤,外显率高,基因位于染色体17q11.2。患病率为3/10万;nfⅱ又称中枢神经纤维瘤或双侧听神经瘤病,基因位于染色体22q。
诊断
(一)辅助检查:x线平片可见各种骨骼畸形;椎管造影、ct及mri可发现中枢神经肿瘤。脑干听觉诱发电位对听神经瘤有较大诊断价值。基因分析可确定nfi和nfⅱ类型。 (二)诊断及鉴别诊断: 1、诊断: ①nf i诊断标准(美国nih,1987):6个或6个以上牛奶咖啡斑,青春期前最大直径>5mm,青春期后>15mm;腋窝和腹股沟区雀斑;2个或2个以上神经纤维瘤或丛状神经纤维瘤;一级亲属中有nf i患者;2个或2以上lisch结节;骨损害; ②nf ⅱ诊断标准:影像学确诊双侧听神经瘤,一级亲属患nfⅱ伴一侧听神经瘤,或伴神经纤维瘤、脑脊膜瘤、胶质瘤、schwann细胞瘤中的两种,青少年后囊下晶状体浑浊。 2、鉴别诊断: 应与结节性硬化、脊髓空洞症、骨纤维结构不良综合征局部软组织蔓状血管瘤鉴别。
治疗
目前无特效治疗。听神经瘤、视神经瘤等颅内及椎管内肿瘤可手术治疗,部分患者可用放疗,癫痫发作者可用抗癫痫药治疗。
并发症
1、皮肤症状: ①几乎所有病例出生时可见皮肤牛奶咖啡斑,形状大小不一,边缘不整,不凸出皮面好发于躯干非暴露部位;青春期前6个以上>5mm皮肤牛奶咖啡斑(青春期后>15mm)具有高度诊断价值,全身和腋窝雀斑也是特征之一。 ②大而黑的色素沉着提示簇状神经纤维瘤,位于中线提示脊髓肿瘤; ③皮肤纤维瘤和纤维软瘤在儿童期发病,主要分布于躯干和面部皮肤,也见于四肢,多呈粉红色,数目不定,可多大数千、大小不等,多为芝麻、绿豆至柑桔大小,质软;软瘤固定或有蒂,触之柔软而有弹性;浅表皮神经的神经纤维瘤似珠样结节,可移动,可引起疼痛、压痛、放射痛或感觉异常;丛状神经纤维瘤是神经干及其分支弥漫性神经纤维瘤,常伴皮肤和皮下组织大量增生,引起该区域或肢体弥漫性肥大,称神经纤维瘤性象皮病。 2、神经症状:约50%的患者出现神经系统症状,主要由中枢周围神经肿瘤压迫引起,其次为胶质细胞增生、血管增生和骨骼畸形所致。 ①颅内肿瘤:听神经瘤最常见,双侧神经瘤是nfⅱ的主要特征,常合并脑膜脊膜瘤、多发性脑膜瘤、神经胶质瘤、脑室管膜瘤、脑膜膨出及脑积水、脊神经后根神经鞘瘤等,视神经、三叉神经及后组脑神经均可发生,少数病例可有智能减退、记忆障碍及痫发作等; ②椎管内肿瘤:脊髓任何平面均可发生单个或多个神经纤维瘤、脊膜瘤,可合并脊柱畸形、脊髓膨胀出去和脊髓空洞症; ③周围神经肿瘤:周围神经均可累及,马尾好发,肿瘤呈串珠状沿神经干分布,一般无明显症状,如突然长大或剧烈疼痛可能为恶变。 3、眼部症状:上睑可见纤维软瘤或丛状神经纤维瘤,眼眶可扪及肿块和突眼搏动,裂隙灯光可见虹膜粟粒橙黄色圆形小结节,为错构瘤,也称lisch结节,可随年龄增大而增多。是nfi特有的表现。眼底可见灰白色肿瘤,视乳头前凸;视神经胶质瘤子可致突眼和视力丧失。 4、常见的先天性骨发育异常包括脊柱侧突、前突和后凸畸形颅骨不对称、缺损和凹陷等。肿瘤直接压迫可导致骨骼改变,如听神经瘤引起内听道扩大,脊神经瘤引起椎间扩大、骨质破坏;长骨、面骨和胸骨过度生长、长骨骨质增生、骨干弯曲和假关节形成也较常见;肾上腺、心、肺、消化道及纵隔等均<
作者: pp 时间: 2008-7-26 06:07
标题: 感谢yangyudong333 战友的精彩病例
感谢yangyudong333 战友的精彩病例!
如果有病史那再也好不过!能提供吗!?
不知能够诊断神经纤维瘤病?
能够找到神经纤维瘤病的诊断依据吗?





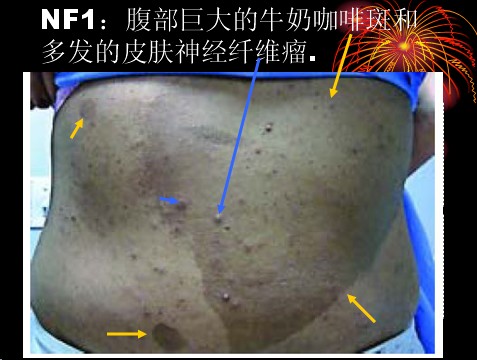
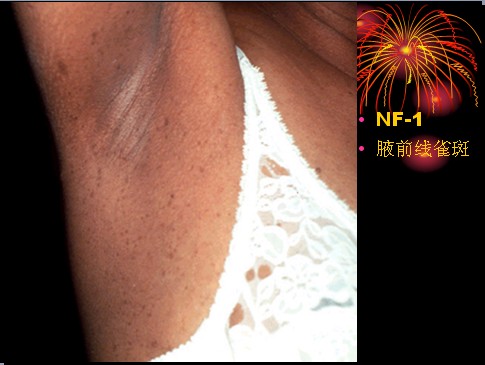

已收录到“影像文摘”中
[本贴已被 翁志蓬 于 2008-8-1 1:22:57 修改过]
作者: pp 时间: 2008-7-26 06:14
标题: 感谢yangyudong333 战友的精彩病例
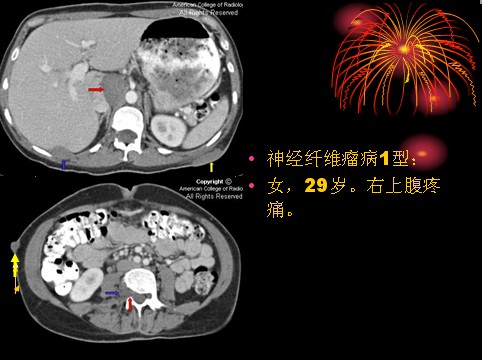
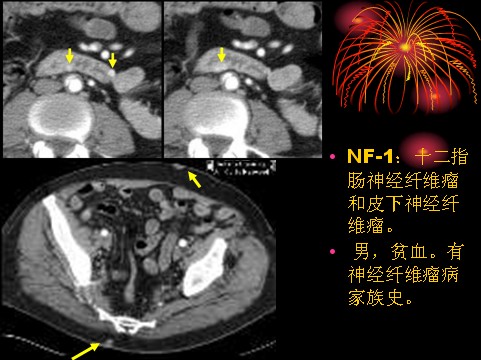
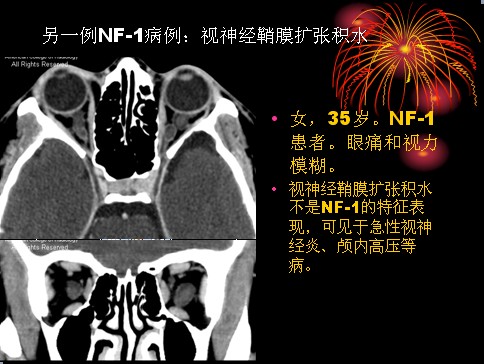


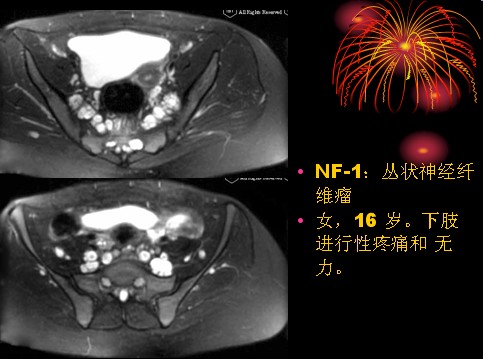
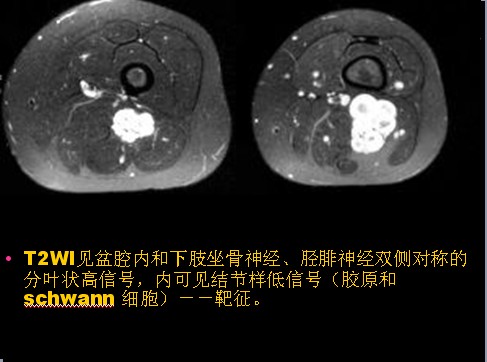
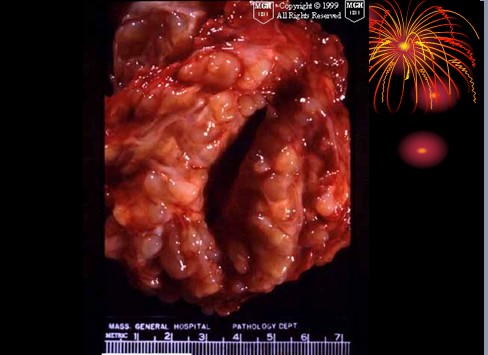


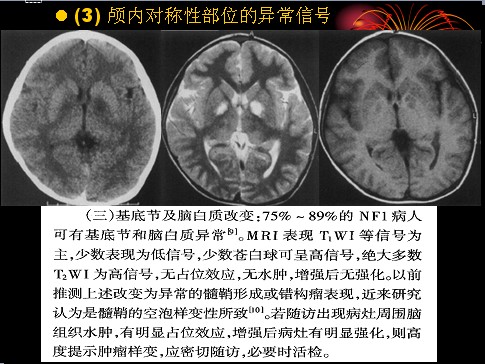

已收录到“影像文摘”中
[本贴已被 翁志蓬 于 2008-8-1 1:20:58 修改过]
作者: aosangwa 时间: 2008-7-26 07:38
好片,支持
作者: dyqct 时间: 2008-7-26 18:22
太精典了!
作者: jinguoji 时间: 2008-7-26 23:58
考虑神经纤维瘤病。
作者: hexue 时间: 2008-8-1 07:02
谢谢楼主,也谢谢各位老师精彩的讲评。确实经典
作者: yuhongjun 时间: 2009-11-23 14:38
符合多发性神经纤维瘤病。
很经典啊。
作者: MZJCTMR 时间: 2010-2-23 01:08
感谢楼主老师的好病例和各位老师的精彩演讲及幻灯。
作者: pujunzhi 时间: 2012-6-17 08:21
感谢楼主老师的好病例和各位老师的精彩演讲及幻灯。学习了!
作者: shibing 时间: 2012-8-22 10:24
多发性神经纤维瘤
| 欢迎光临 医影在线 (http://bbs.radida.com/bbs/) |
Powered by Discuz! X3.2 |