该病例为典型的成人型多囊肝、多囊肾。左侧肾中上极包膜下的致密影考虑小结石。
成人型多囊肝多囊肾的起源及特点 肝囊肿合并肾囊肿也称之为肝肾多囊病,属于先天性疾病,该病是常染色体显性遗传性疾病。在本病的起源上多数作者认为多囊肝是在多囊肾的基础上基因突变的结果,并定位于16号染色体上,也有学者认为该病由于胚胎发育不良所致,于胚胎早期管道(包括肝小叶内胆小管)形成时排列失常,造成无数迷管,于胎胚晚期未退化,且逐渐扩张成囊,成多发性囊肿病。根据发病年龄的不同,将其分为儿童型和成人型。本组病例均为成人型。本病多发生于双侧肾脏,单侧肾脏少见,本病可继发感染(50%-75%)、结石(10%)以及囊内出血和囊壁的钙化,也可以恶变,并有阻塞性黄疸及合并皮下囊肿的报道。临床特点病人早期无症状,就诊时肾脏功能已经明显受损,肝功能受损程度较轻.
多囊肝多囊肾的ct、mr表现与病理表现的相关性 从病例的术中、术后病理观察可以发现,肝囊肿内的液体较为清亮,呈水样,压力较高,在ct和mr上表现为密度及信号的均匀一致相吻合,正常肝脏组织受压变薄形成假包膜并伴有一定的纤维化,ct增强上表现为囊肿的边缘可见轻度强化的表现,未发现肝脏囊肿感染和出血的表现。而肾囊肿的情况较为复杂,开窗减压术中所见的囊内容物成分不一致,可为淡黄色、脓性黄褐色、巧克力色、暗绿色液体,这种囊内容物的出现可能与慢性肾囊肿出血及感染有关,造成囊肿ct值的差异其内密度不均匀,肾脏囊肿也有假包膜征象,囊壁较薄,由纤维组织构成,部分较厚的囊壁为受压变薄的肾组织,所以可见部分囊壁强化.
[本贴已被 xiaoniu 于 2006-11-5 17:48:59 修改过] | 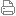







 发表于 2006-11-4 17:32
发表于 2006-11-4 17:32
 QQ好友和群
QQ好友和群 QQ空间
QQ空间 腾讯微博
腾讯微博 腾讯朋友
腾讯朋友 收藏
收藏 赞同
赞同 反对
反对